India has established itself as one of the world’s most important destinations for pharmaceutical clinical research — combining a large, genetically diverse patient population, world-class research institutions, experienced clinical investigators, and a regulatory framework that has progressively aligned with international clinical trial standards. Understanding how clinical trials and drug approval India process works is essential knowledge for pharmaceutical companies developing new medicines, generic drug manufacturers seeking market authorization, international investors evaluating Indian pharmaceutical development programs, and global healthcare buyers who want to understand the regulatory foundation underlying Indian pharmaceutical products they source.
India’s Pharmaceutical Regulatory Architecture
Regulatory framework pharmaceuticals India is administered primarily through two interconnected authorities whose respective responsibilities define the complete drug approval landscape:
Central Drugs Standard Control Organisation (CDSCO) — India’s national pharmaceutical regulatory authority — functions under the Ministry of Health and Family Welfare and is responsible for approving new drugs, overseeing clinical trial conduct, evaluating drug applications, and setting national pharmaceutical quality and safety standards. CDSCO operates through a central headquarters in New Delhi with zonal and sub-zonal offices across India that manage specific regulatory functions.
Drug Controller General of India (DCGI) — the head of CDSCO — holds ultimate authority over DCGI approval drugs India process decisions, providing the final regulatory authorization for new drug approvals, clinical trial permissions, and import licenses for pharmaceutical products entering the Indian market.
State Drug Authorities — complement CDSCO’s central functions by regulating pharmaceutical manufacturing through manufacturing license issuance and manufacturing site GMP inspections — creating a two-tier regulatory system where central authority governs product approvals while state authorities govern manufacturing compliance.
The New Drugs and Clinical Trials Rules 2019: The Current Framework
Clinical research guidelines India were substantially updated through the New Drugs and Clinical Trials Rules 2019 — a comprehensive regulatory reform that replaced the clinical trial provisions of the Drugs and Cosmetics Act with a modernized framework addressing clinical trial conduct, new drug approvals, bioequivalence studies, and the ethics committee oversight that protects clinical trial participants.
Indian clinical trials regulations under NDCT Rules 2019 introduced several significant improvements over the previous framework:
Accelerated timelines — defined maximum timelines for CDSCO review of clinical trial applications — 30 working days for review of new clinical trial applications — creating regulatory predictability that previous open-ended review timelines did not provide.
Global trial harmonization — provisions facilitating simultaneous global clinical trials — including innovative drugs being developed simultaneously in India and internationally — reducing the delays that previously required sequential Indian trial conduct after international trial completion.
Waiver provisions — CDSCO may waive Indian clinical trial requirements for new drugs already approved by stringent regulatory authorities in specified countries — allowing Indian market approval based on foreign clinical data for medicines with established international regulatory standing.
Compensation framework — defined compensation provisions for clinical trial participants experiencing adverse events — addressing the participant protection concerns that previous regulatory gaps had created.
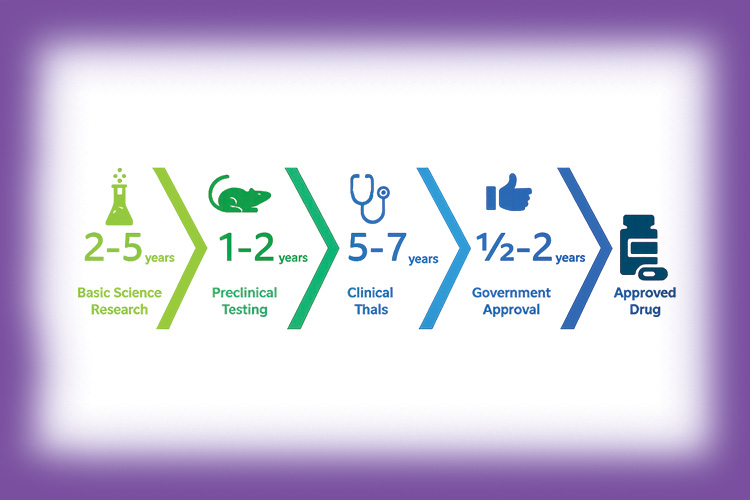
Phases of Clinical Trials in India
Phases of clinical trials India follow the internationally recognized four-phase clinical development framework — with each phase building progressively on the safety and efficacy evidence established in prior phases:
Phase I — First-in-Human Safety Studies — conducted in small groups of healthy volunteers or, for some therapeutic areas including oncology, in patients. Phase I trials establish human pharmacokinetics, dose escalation safety, and the maximum tolerated dose that subsequent phases will study. CDSCO clinical trial process India for Phase I requires ethics committee approval and CDSCO clinical trial permission before any human administration can begin.
Phase II — Proof of Concept and Dose Finding — conducted in small patient populations to establish therapeutic dose ranges, preliminary efficacy signals, and safety profiles in the target patient population. Phase II trials generate the evidence base that informs Phase III trial design — determining the doses, patient populations, and efficacy endpoints that the pivotal Phase III trials will evaluate.
Phase III — Pivotal Efficacy and Safety Studies — the large-scale, randomized, controlled trials that provide the definitive evidence of therapeutic efficacy and safety required for regulatory approval. Phase III trials enroll hundreds to thousands of patients across multiple clinical sites — generating the statistical power needed to demonstrate clinically meaningful efficacy with acceptable safety profiles. Phase III data forms the primary basis for new drug approval steps India evaluation.
Phase IV — Post-Marketing Surveillance — conducted after drug approval to monitor real-world safety and efficacy in broad patient populations — identifying adverse events, drug interactions, and safety signals that pre-approval trials with selected patient populations may not have detected. Phase IV studies fulfill ongoing pharmacovigilance obligations and may support label expansions for approved drugs.
Ethics Committee Oversight: Protecting Trial Participants
Ethics committee clinical trials India oversight represents one of the most important participant protection mechanisms in India’s clinical trial regulatory framework — with independent ethics committees providing review of clinical trial protocols, informed consent processes, and ongoing trial conduct at every clinical trial site.
NDCT Rules 2019 significantly strengthened ethics committee requirements — establishing registration requirements for ethics committees conducting clinical trial oversight, defining minimum composition requirements ensuring independent and multidisciplinary membership, and creating accountability frameworks that professionalize ethics committee functions that were previously more variable in their rigor.
Key ethics committee responsibilities in drug approval process India pharma oversight include:
Protocol scientific and ethical review — evaluating whether trial design is scientifically sound, whether risks to participants are justified by potential benefits, and whether the trial addresses genuine scientific questions rather than simply fulfilling regulatory requirements.
Informed consent process approval — reviewing and approving the informed consent documents and processes that ensure trial participants receive complete, comprehensible information about trial participation before providing voluntary consent.
Ongoing safety monitoring — reviewing serious adverse event reports and data safety monitoring committee recommendations throughout trial conduct — with authority to require protocol modifications or trial suspension when participant safety concerns arise.
New Drug Approval Steps India: The Complete Pathway
New drug approval steps India for original new chemical entities follow a comprehensive development and regulatory submission pathway:
Investigational New Drug Application — the initial regulatory submission requesting CDSCO permission to begin clinical trials in India — including preclinical pharmacology and toxicology data, pharmaceutical quality information, proposed clinical trial protocol, investigator qualifications, and ethics committee approval documentation.
Clinical Development Program — execution of Phase I through Phase III clinical trials under CDSCO-approved protocols — generating the safety and efficacy evidence that supports the new drug application.
New Drug Application submission — comprehensive technical dossier submission to CDSCO including Module 1 administrative information, Module 2 summaries, Module 3 pharmaceutical quality data, Module 4 preclinical data, and Module 5 clinical data — prepared in CTD format aligned with ICH guidelines.
CDSCO technical review — expert committee scientific evaluation of the NDA submission — with Subject Expert Committee review providing specialized scientific assessment in relevant therapeutic areas.
DCGI approval decision — final regulatory decision by the Drug Controller General of India — granting approval, requesting additional data, or declining approval with stated reasons — following technical committee recommendation.
Generic Drug Approval: The Bioequivalence Pathway
Drug approval process India pharma for generic medicines follows a distinct but equally rigorous pathway — focused on demonstrating bioequivalence to the reference listed drug rather than repeating the complete clinical development program that established the originator medicine’s efficacy and safety.
Pharma regulatory compliance India bioequivalence requirements for generic drug approval include:
Pharmaceutical equivalence — demonstrating that the generic medicine contains the same active pharmaceutical ingredient, in the same dosage form and strength, with the same route of administration as the reference product.
Bioequivalence study conduct — pharmacokinetic studies in healthy volunteers — typically crossover design studies comparing blood concentration profiles of the generic and reference products — demonstrating that 90% confidence intervals for AUC and Cmax ratios fall within the 80-125% regulatory acceptance range.
Pharmaceutical quality documentation — complete quality module including formulation details, manufacturing process, analytical methods, specifications, and stability data under ICH climatic zone conditions.
Labeling compliance — product labeling meeting CDSCO requirements for generic drug products — including complete prescribing information, contraindication warnings, and the regulatory statements required for Indian market pharmaceutical products.
India’s Clinical Trial Industry: The Research Ecosystem
Clinical research guidelines India have supported the development of a substantial clinical research industry — with India offering pharmaceutical companies conducting global clinical trials several distinctive advantages:
Patient population diversity — India’s large, genetically diverse population provides access to patient cohorts representing diverse genetic backgrounds, disease presentations, and treatment histories that strengthen the global relevance of clinical evidence generated in Indian trials.
Disease burden breadth — India’s significant burden of both communicable and non-communicable diseases provides clinical trial access to patient populations with conditions ranging from tuberculosis and malaria through cardiovascular diseases, diabetes, and oncology that represent major global clinical development priorities.
Experienced clinical investigator base — India has developed a substantial community of clinical investigators with international clinical trial training, GCP compliance experience, and the scientific expertise to conduct complex multi-center trials meeting ICH Good Clinical Practice standards that regulatory authorities in major markets require.
Cost efficiency — clinical trial conduct costs in India are significantly lower than comparable trial execution in Western markets — enabling pharmaceutical companies to conduct high-quality clinical research within development budgets that comparable Western trial costs would not accommodate.
Onco India International: Built on India’s Regulatory Excellence
At Onco India International, our pharmaceutical manufacturing operations are grounded in the same regulatory framework that governs clinical trials and drug approval India — with our products approved through CDSCO regulatory processes that ensure pharmaceutical quality, safety, and efficacy standards are independently verified before any medicine reaches the patients who depend on it.
Our regulatory affairs team maintains the deep understanding of India’s pharmaceutical regulatory framework that ensures every product we manufacture and export is supported by the regulatory documentation, quality evidence, and compliance standing that international buyers require from their Indian pharmaceutical supply partners.
Contact Onco India International today to discuss your pharmaceutical supply requirements and experience the regulatory expertise, manufacturing quality, and genuine compliance commitment that defines Onco India International as a trusted Indian pharmaceutical manufacturing and export partner.